II. BASIC CONCEPTS AND TERMINOLOGY
Distant similarities: an introduction
Pattern terminology: confusing names to simple things
Sequences are a kind of chemical structures or texts
Similarity of two sequences: alignments
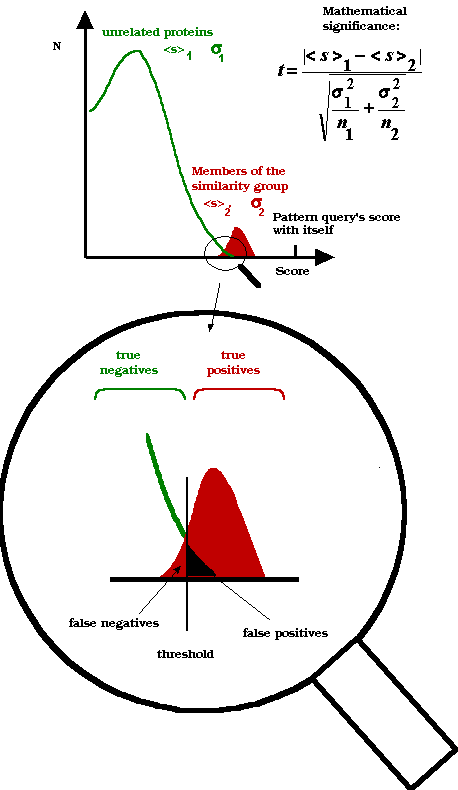
Biological significance, mathematical significance
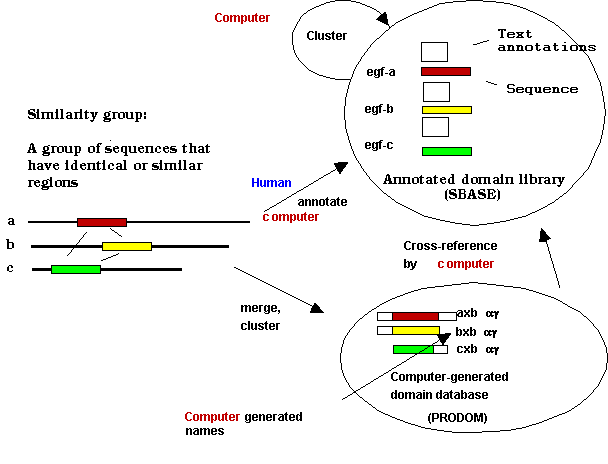
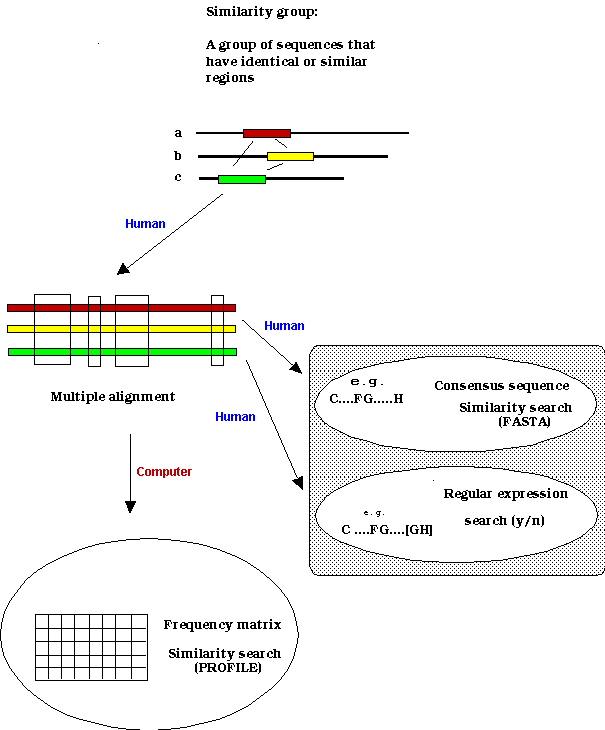
Similarity groups:patterns, motifs, signatures
Patterns as representations of similarity groups
Profiles as representations of similarity groups
Distant similarities:
an introduction
Database search is probably the most widely used approach to predict the biological function of a newly determined protein sequence. The likelihood of finding homologues (i.e. evolutionarily related proteins) to a sequence is currently higher than 80% for bacteria; 70% for yeast and perhaps 60% for animal sequence queries.The rest are truely novel proteins, but an estimated half of them are non-trivial cases that can be identified only by more sophisticated methods of analysis. We refer to these cases as "distant similarities".
If two large proteins share only a few common domains, or the level
of identity is around 25% or below, detection of similarity often becomes
difficult. First, random identities present in the alignment may
"mask"
the biologically important sequence patterns. Second, search programs such
as FASTA, FASTDB or BLAST give only the best homology regions between the
query and a database entry, so weaker homologies between the query and
the same entry can remain undetected. In this "twighlight zone"
of homologies (Rus Doolittle's expression), biological significance does
not coincide with mathematical significance, so simple similarity searches
do not give unequivocal results. In these cases, the experimenter is often
tempted to give up saying "the sequence showed no significant homology
to any other sequence in the database". In this chapter we describe
a number of simple possibilities to analyze a newly determined protein
sequence that you may try in order to go beyond the scope of simple database
search.
There are no general solutions for finding distant homologies. However,
there are a number of useful solutions that are likely to work with various
problems, so it is a good idea to try a number of different programs and
databases. ICGEBnet maintains a growing collection of these and useful
WWW-links are provided in the last chapter of this tutorial.
Pattern terminology: confusing names to simple things
When discussing sequence homology we use terms as "patterns", "motifs" and "similarity". These are broad and vague terms, it is enough to think about concepts like patterns of behaviour or musical motifs. When we use these terms for sequences, we use them in a given context and a restricted sense. Typically, a PROSITE pattern (see below) is a special sequence pattern that makes sense only in the context of the PROSITE database and the software that can be used to search it. In other terms, there is a proper mathematical definition of patterns used in PROSITE and in the search software. If we keep this in mind we will not be disturbed by the fact that some workers call the PROSITE patterns as "motifs" - they refer to the same thing. This kind of problem is characteristic of every new field in science.
If we still want to make a distinction, the word "motif" is
used for simpler patterns of identity that are usually short and can be
described in simple terms. The word "pattern" itself is more
used to more detailed descriptions, typically for those comprising several
motifs. However, it is also correct to speak about a "hydrophobicity
pattern", i.e. the distribution of a quantitative property along the
sequence. Finally, the word "profile" is used mostly for complex,
quantitative descriptions derived from multiple alignment.
Sequences are a kind of chemical structures or texts
Chemical structures are collections of elements (typically atoms) with connections (like chemical bonds) between them. Protein sequences are composed of a limited set of special elements, amino acid residues, and only one type of connection is represented, that of the protein backbone which only shows which residues are next to each other (this connection can be also termed "sequential vicinity"). This representation is very similar to texts, especially if we use the one-letter code for amino acids. In fact, most mathematical methods used for sequences originate from text processing. Enough to think about concepts such as "deletions" and "insertions" now widely used in genetics which also come from text editing.
So we have two metaphors for sequences: chemical structures and
texts. When to use these? Sequence analysis, such as database searching,
can be much better understood with the text metaphor. This is valid to
all problems where we keep the amino acid residue representation as standard.
If we need more versatile descriptions, the chemical structure metaphor
is more useful. For example, the structure of multidomain proteins can
be described as a series of complex elements, such as signal peptides,
immunoglobulin-domains etc. The chemical structure analogy is especially
conspicuous when dealing with the 3D structure of proteins composed of
a few a-helices and b-sheets
in a special arrangement.
Similarity of two sequences: alignments
The concept of similarity is basically a very complex notion. For sequences we use a very restricted definition to which a mathematical concept can be assigned. The similarity of two sequences is best defined by the alignment of the two sequences, and can be characterized by the alignment pattern and the similarity score.
There are many ways to align sequences, and by "alignment"
we refer to the best alignment that has the maximum number of identical
residues.![]() By alignment pattern we refer to the pattern of identical residues as shown
in the following example:
By alignment pattern we refer to the pattern of identical residues as shown
in the following example: