C . . . H . . . C
Is this pattern important?
The first thing to do is a multiple alignment using the three candidate
sequences. In this multiple alignment we may discover new sequence identities
that we may have overlooked previously, e.g. we discover that the shared
pattern is
C . . . H . . . C . . C
(Some of you may have noticed that this example is the pattern in zinc-fingers.)
But is this pattern important???
First we have to look at the pattern using the rules described above
(frequent residues, repetitive sequences, structurally important residues).
This example is seemingly OK, we have cysteines (perhaps structurally important)
plus a histidine which is not extremely frequent.
Second, we have to ascertain, if this pattern is not present in any
other sequences. So we test out database of 10 and find that it is in fact
absent from the other sequences.
Third, and most important, we have to find out whether or not this pattern
can have a biological meaning at all. If all three sequences are of the
same function, e.g. DNA-binding proteins, on can think that the pattern
has to do with the function. But let's not be overconfident. Leucine-zippers
are parts of DNA-binding proteins, but they mediate dimerization and are
not directly involved in binding DNA. So, it is not possible to firmly
establish a biological function in most cases.
The solution can be "statistical": If we have a "sufficiently
big" number of examples in which a biological function is associated
with a certain sequence pattern, we can feel encouraged. It is equally
important that the pattern should not be found in those sequences which
do not carry the same biological function. In a different case, we may
also have strong biological clues. For example we may know that 3 protein
sequences are involved in the same biological function in different organisms.
In this case, the group is given in advance and we are looking for a sequence
pattern that may be used to characterize the group, and moreover, may be
responsible in the biological function.
Let's say we compare our above pattern to two groups of sequences, i)
DNA-binding proteins and ii) non-DNA-binding proteins. We find that the
DNA-binding proteins always have a score greater than 10 and the non-DNA-binding
proteins have scores less than 5. We are now ready with the rule: any sequence
producing a similarity score greater than, say, 7 in comparison with the
pattern, may belong to the group of DNA-binding proteins. In other terms,
we define a threshold value. This rule may work, but in real life situations
we usually have complications, like:
i) DNA-binding proteins that score below 7. We call these "false
negatives" because they scored negative in the test but are in fact
positive.
ii) Non-DNA-binding proteins that score above 7. We call these 'false
positives" since they scored positive but are in fact negative.
In fact, for a "perfect pattern" we require that it should
occur only in the "true positive" sequences (that really carry
the function in question), and in none of the "true-negative"
sequences (which do not carry the function). Also, we do not want "false
positives" and "false negatives".
Real-life situations are unfortunately even more complicated. usually
we have a small group (a "test group") of sequences of which
we suspect they may carry a common function. We establish a pattern and
then use some rudimentary procedure to calculate a "similarity score"
for that pattern. We use the entire database as the other group. Using
the Student principle for comparing two groups (characterized each by an
average and standard deviation), one can calculate the statistical significance
of the difference:
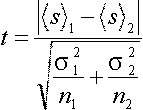
If this value if big, the separation is "mathematically significant",
so we can trust the pattern better. It is easier to depict graphically
the situation, as shown in figure
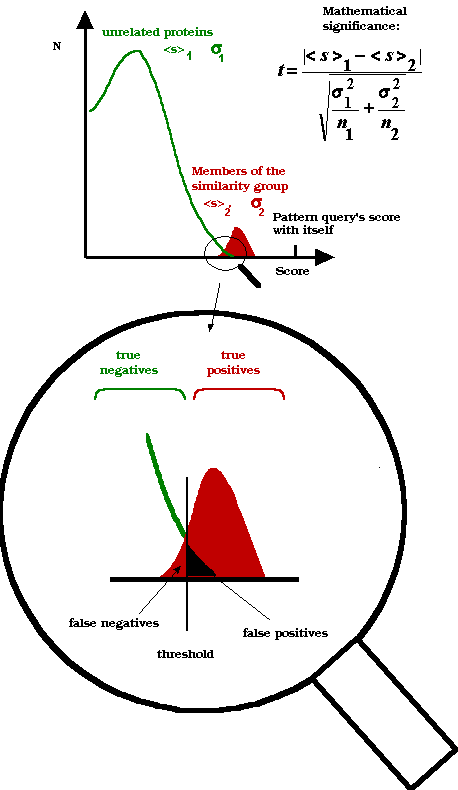{kind=link}