
NCBI ICGEB Problem Set
Entrez
Back to top↑- Use the search GenBank box on the NCBI homepage with accession number U00089 to retrieve the CON division record for the complete Mycoplasma pneumoniae complete genome. How many GenBank records make up this record? Use the display pull-down list to change to the graphic view to see graphically how these segments map and features map onto the sequence.
- Retrieve the draft record AC013402. How many unordered pieces are in the record now? How many times has it been updated since it first appeared? Trace the history all the way back to the first version. Based on the update date when did this record first appear how many unordered pieces were there then? Now use electronic PCR (linked as a “hotspot” on the NCBI homepage to identify STS markers present in this record. How many are there? These include radiation hybrid and genetic markers. Which one is also a genetic marker?
- The Entrez Properties field stores information about the kind of sequence and its source. You can use the index feature on the Preview/Index tab to display the terms that are indexed for this field. Use the gbdiv sets of terms to count the number of records currently the HTG, GSS, EST, HTC divisions (e.g. gbdiv est[Properties]). Use the molecule type term biomol to count the number of mRNA and genomic DNA records in Entrez nucleotides (e.g. biomol mrna[Properties]).
- Devise a query to retrieve the ten largest human genomic sequences. (You may specify this as “Sequence Length” range with the second endpoint as some arbitrarily large number e.g. 999999999). The largest of these, those with NT_xxxxx style accession number will be RefSeq contigs from the human genome project data. Now find the ten largest single (not contig records) human sequences in the nucleotide database. You can do this by “NOTing” out the RefSeq records using the srcdb terms of the Properties field. Now find the largest finished single human record.
- Use the program BLAST 2 Sequences to compare the RefSeq mRNA for CFTR (NM_000492) with the model transcript (XM_004980) predicted from the human genome. Are there any mismatches? Now compare the original GenBank sequence for the CFTR mRNA (M28668) to the RefSeq and note any differences in sequence.
- Use Entrez nucleotides to find the full-length cDNA (mRNA) sequence for Plasmodium falciparum glyceraldehyde 3 phosphate dehydrogenase (GAPD). This time start by typing Plasmodium in the search box without limiting to any field. How many records do you retrieve? Browse through your results to find some records that are not from Plasmodium. Display a few of these to see why you retrieved them; you should find “Plasmodium” somewhere on the record. Now use the Limits tab to restrict to Plasmodium in the Organism field [Organism]. How many nucleotide records in Entrez are from Plasmodium? Now find GAPD records by using the Preview/Index tab to add glyceraldehyde 3 phosphate dehydrogenase as a Title Word term. How many records did you retrieve?
- Search for population and phylogenetic studies on the mammalian order carnivora in Entrez PopSet. Find the study on brown bears and polar bears and display the alignment. What gene or molecular regions were used in this study? Use the toolbar link to display variations in the alignment. Are there fixed differences in the sequences from the brown bear, Ursus arctos, and the polar bear sequences in the alignment? How about if the Ursus arctos sequence from the “ABC” islands islands (Sequence 7) is removed. Link to the article to read more about these remarkable results.
Go back to the original set of carnivora PopSets.
- Substantial EST data are available for two species of filarial nematodes that are human parasites. Use the Taxonomy Browser to examine the number of nucleotide sequences for the superfamily Filaroidea and determine which two species these are. How many nucleotide and protein sequences are there for each of these two species? Examine nucleotide records for each of these and find which laboratory is producing these ESTs.
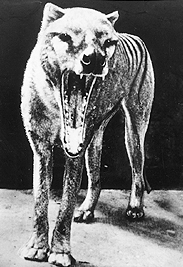
- The last known Tasmanian tiger died in the Hobart Zoo in 1936. DNA sequences have been obtained from museum specimens. You can retrieve Tasmanian tiger sequences using the Taxonomy Browser. Search the taxonomy database for Tasmanian Tiger. How many DNA and protein sequences are there? What genes were cloned? You can build a phylogenetic data set that could be used to analyze the taxonomic position of the Tasmanian Tiger with the Taxonomy Browser. Click on the Metatheria (Marsupial) link in the lineage of the tiger. How many nucleotide sequences are there for Metatheria? Retrieve the entry for Metatheria and get the nucleotide sequences. In Entrez you can refine the query to include only cytochrome b sequences through the Preview/Index tab. How many marsupial cytochrome b sequences are there? You could save these in FASTA format for use in the phylogenetic analysis if you wanted. You could browse up the lineage further to get an outgroup sequence
Structures
Back to top↑- Use Entrez Structures to retrieve the X-ray crystal structure of the oxidized form of bovine cytochrome B5 (1CYO). View the structure with Cn3D. Locate the iron atom in the heme complex. Which two amino acids coordinate with the iron? Align the yeast cytochrome B5 sequence (accession number P40312) using the “Download Sequence” option on the “Align” menu on the sequence view window. You will first have to configure Cn3D as a network client. Do this by selecting Net Configure from the “Options” menu of the structure viewer. Click the normal radio button and click “Accept”, then re-start Cn3D.
- Use Entrez structures to retrieve the structure record for the human VH-1-related phosphatase (1VHR). View the structure with Cn3D. How many chains are in the asymmetric unit of this crystal structure? How many alpha helices and beta strands are present in each of the subunits? Close out that invocation of Cn3d. Find structure neighbors for 1VHR chain A. How much identity does the phosphatase from Yersinia have with 1VHR_A? Display the alignment of these two proteins with Cn3d. Select all atoms and all display only aligned chains. Note that the sulfate ion bound by 1YTS is now nearly superimposed on the phosphate of the substrate analog in 1VHR. Zoom into this region. (Click Ctrl and drag the mouse.) There is a conserved catalytic nucleophile in the active site of both of the proteins (a serine in one and a cysteine in the other). Identify the nucleophile by highlighting the cysteines in the alignment and displaying side chains in the viewer.
Problems
Back to top↑- Michael Crichton’s fantasy about cloning dinosaurs, Jurassic Park contains a putative dinosaur DNA sequence. Use nucleotide-nucleotide BLAST against the default nucleotide database to identify the real source of the following sequence:
DinoDNA "Dinosaur DNA" from Crichton's JURASSIC PARK p. 103 nt 1-1200 GCGTTGCTGGCGTTTTTCCATAGGCTCCGCCCCCCTGACGAGCATCACAAAAATCGACGC GGTGGCGAAACCCGACAGGACTATAAAGATACCAGGCGTTTCCCCCTGGAAGCTCCCTCG TGTTCCGACCCTGCCGCTTACCGGATACCTGTCCGCCTTTCTCCCTTCGGGAAGCGTGGC TGCTCACGCTGTACCTATCTCAGTTCGGTGTAGGTCGTTCGCTCCAAGCTGGGCTGTGTG CCGTTCAGCCCGACCGCTGCGCCTTATCCGGTAACTATCGTCTTGAGTCCAACCCGGTAA AGTAGGACAGGTGCCGGCAGCGCTCTGGGTCATTTTCGGCGAGGACCGCTTTCGCTGGAG ATCGGCCTGTCGCTTGCGGTATTCGGAATCTTGCACGCCCTCGCTCAAGCCTTCGTCACT CCAAACGTTTCGGCGAGAAGCAGGCCATTATCGCCGGCATGGCGGCCGACGCGCTGGGCT GGCGTTCGCGACGCGAGGCTGGATGGCCTTCCCCATTATGATTCTTCTCGCTTCCGGCGG CCCGCGTTGCAGGCCATGCTGTCCAGGCAGGTAGATGACGACCATCAGGGACAGCTTCAA CGGCTCTTACCAGCCTAACTTCGATCACTGGACCGCTGATCGTCACGGCGATTTATGCCG CACATGGACGCGTTGCTGGCGTTTTTCCATAGGCTCCGCCCCCCTGACGAGCATCACAAA CAAGTCAGAGGTGGCGAAACCCGACAGGACTATAAAGATACCAGGCGTTTCCCCCTGGAA GCGCTCTCCTGTTCCGACCCTGCCGCTTACCGGATACCTGTCCGCCTTTCTCCCTTCGGG CTTTCTCAATGCTCACGCTGTAGGTATCTCAGTTCGGTGTAGGTCGTTCGCTCCAAGCTG ACGAACCCCCCGTTCAGCCCGACCGCTGCGCCTTATCCGGTAACTATCGTCTTGAGTCCA ACACGACTTAACGGGTTGGCATGGATTGTAGGCGCCGCCCTATACCTTGTCTGCCTCCCC GCGGTGCATGGAGCCGGGCCACCTCGACCTGAATGGAAGCCGGCGGCACCTCGCTAACGG CCAAGAATTGGAGCCAATCAATTCTTGCGGAGAACTGTGAATGCGCAAACCAACCCTTGG CCATCGCGTCCGCCATCTCCAGCAGCCGCACGCGGCGCATCTCGGGCAGCGTTGGGTCCT
Mark Boguski of the NBCI noticed this and supplied Crichton with a better sequence for the sequel, The Lost World. Identify the most likely source of this sequence using the nucleotide-nucleotide BLAST. Mark embedded his name in the sequence he provided. To see Mark’s name use the translating BLAST (blastx) page the sequence below. (Look for MARK WAS HERE NIH).
DinoDNA "Dinosaur DNA" from Crichton's THE LOST WORLD p. 135 GAATTCCGGAAGCGAGCAAGAGATAAGTCCTGGCATCAGATACAGTTGGAGATAAGGACG GACGTGTGGCAGCTCCCGCAGAGGATTCACTGGAAGTGCATTACCTATCCCATGGGAGCC ATGGAGTTCGTGGCGCTGGGGGGGCCGGATGCGGGCTCCCCCACTCCGTTCCCTGATGAA GCCGGAGCCTTCCTGGGGCTGGGGGGGGGCGAGAGGACGGAGGCGGGGGGGCTGCTGGCC TCCTACCCCCCCTCAGGCCGCGTGTCCCTGGTGCCGTGGGCAGACACGGGTACTTTGGGG ACCCCCCAGTGGGTGCCGCCCGCCACCCAAATGGAGCCCCCCCACTACCTGGAGCTGCTG CAACCCCCCCGGGGCAGCCCCCCCCATCCCTCCTCCGGGCCCCTACTGCCACTCAGCAGC GGGCCCCCACCCTGCGAGGCCCGTGAGTGCGTCATGGCCAGGAAGAACTGCGGAGCGACG GCAACGCCGCTGTGGCGCCGGGACGGCACCGGGCATTACCTGTGCAACTGGGCCTCAGCC TGCGGGCTCTACCACCGCCTCAACGGCCAGAACCGCCCGCTCATCCGCCCCAAAAAGCGC CTGCTGGTGAGTAAGCGCGCAGGCACAGTGTGCAGCCACGAGCGTGAAAACTGCCAGACA TCCACCACCACTCTGTGGCGTCGCAGCCCCATGGGGGACCCCGTCTGCAACAACATTCAC GCCTGCGGCCTCTACTACAAACTGCACCAAGTGAACCGCCCCCTCACGATGCGCAAAGAC GGAATCCAAACCCGAAACCGCAAAGTTTCCTCCAAGGGTAAAAAGCGGCGCCCCCCGGGG GGGGGAAACCCCTCCGCCACCGCGGGAGGGGGCGCTCCTATGGGGGGAGGGGGGGACCCC TCTATGCCCCCCCCGCCGCCCCCCCCGGCCGCCGCCCCCCCTCAAAGCGACGCTCTGTAC GCTCTCGGCCCCGTGGTCCTTTCGGGCCATTTTCTGCCCTTTGGAAACTCCGGAGGGTTT TTTGGGGGGGGGGCGGGGGGTTACACGGCCCCCCCGGGGCTGAGCCCGCAGATTTAAATA ATAACTCTGACGTGGGCAAGTGGGCCTTGCTGAGAAGACAGTGTAACATAATAATTTGCA CCTCGGCAATTGCAGAGGGTCGATCTCCACTTTGGACACAACAGGGCTACTCGGTAGGAC CAGATAAGCACTTTGCTCCCTGGACTGAAAAAGAAAGGATTTATCTGTTTGCTTCTTGCT GACAAATCCCTGTGAAAGGTAAAAGTCGGACACAGCAATCGATTATTTCTCGCCTGTGTG AAATTACTGTGAATATTGTAAATATATATATATATATATATATATCTGTATAGAACAGCC TCGGAGGCGGCATGGACCCAGCGTAGATCATGCTGGATTTGTACTGCCGGAATTC
- Higher eukaryotic genomes contain large amounts of repetitive DNA. The most abundant interspersed repeat in the human genome is the Alu element. Alus tend to occur near genes within the introns or in the regions between genes. In some cases, their presence and absence can fairly accurately show the intron-exon structure of a gene. Demonstrate this by performing a nucleotide-nucleotide BLAST search against the Alu database with the genomic sequence of the human Von Hippel Lindau syndrome gene, accession AF010238. Note that the exons appear in the BLAST graphic as places where the Alu elements do not align.
- The C. elegans gene SMA-4 is a member of the dwarfins gene family, which plays a role in TGF-mediated signal transduction. In order to identify potential homologs in other species, use the protein-protein blast page to perform a search against the non-redundant protein database (nr) using SMA-4 (accession number P45897) as the query sequence. Find all chicken (Gallus gallus) proteins that are similar to SMA-4. ( Use the Tax Blast link at the upper left of the graphic to help in finding the chicken proteins.) Now run the search again and restrict to chicken proteins through the Entrez query advanced option. What proteins are found? Compare the Expectation values of these hits to the same hits found against nr with no organism restriction. Why are the E values different for the same scores and alignments?
- The human fragile histidine triad protein (FHIT) (SWISS-PROT: P49789) has been shown to be structurally homologous to galactose-1-phosphate uridylyltransferase. However, this relationship is not apparent in an ordinary BLAST search. Perform a protein-protein blast search against the SwissProt database with P49789 and search your results for galactose-1-phosphate uridylyltransferases. Now use PSI-BLAST to verify the relationship between these two protein families
- Find the unannotated genomic scaffold for Drosophila melanogaster, AE003584, using Entrez nucleotides. Display protein links to see the predicted proteins for this scaffold. Use the CDD search to identify conserved domains present in the tenth predicted protein (gi: 7295996) and suggest a potential function for this hypothetical protein.
- As the database grows so does the number of chance occurrences of amino acid motifs that spell out words or people’s names in single-letter amino acid codes. One such name motif is ELVIS. Find the number of occurrences of ELVIS in the protein nr. To get any hits at all, you will have to adjust several of the advanced BLAST parameters including the Expect value, Word size, and Score Matrix. Adjust some of these in the “Other advanced options” box. Options are entered command line style. For example, typing
-e 10000sets the Expect value cut-off to 10000. See the BLAST “Frequently Asked Questions” linked on the left sidebar of the BLAST page on “How do I perform a similarity search with a short peptide/nucleotide sequence?” for more information. We now have a page with presets optimized to find short nearly exact matches. You can cheat and run the search on this page to see the correct parameters to use.
Genome Resources Questions
Back to top↑- The Mycobacteria are highly specialized intracellular parasites. They have unusual metabolisms and seem to have acquired genes by horizontal transfer from their host. You can demonstrate these features by comparing the Mycobacterium tuberculosis genome. Use the Entrez genomes page to view the genomes of Escherichia coli and Mycobacterium tuberculosis. (You may want to launch two browsers to do this example.) Display the distribution of BLAST hits by Taxa for each and compare the distribution of homologs. Which organism has more best hits to Eukaryotes? Now display the BLAST hits for each by COGs (clusters of orthologous groups). The tuberculosis organism has a disproportionate portion of the genome devoted to the metabolism of what class of biomolecules?
- mRNA that hybridized to the EST sequence with accession number AI589456 was highly expressed in a human liver tumor sample. Use human UniGene data to identify this gene. Link to LocusLink. What is the function of this protein? Go back to Go back to UniGene. Look at the ESTs in this cluster. How many are there? Identify a pair of ESTs that come from the same clone ID. Use BLAST two sequences to align these to the full-length RefSeq mRNA from the LocusLink entry. Are there any mismatches? Another mRNA hybridizes to AI150058. What information can you find about this gene?
- Retrieve the LocusLink entry for human BRCA1. Scroll down to the NCBI Reference Sequences section. How many splice variants are reported for this gene’s transcripts? Use the sv link on the NCBI contig to see a graphical view that shows these splice variants more clearly. Scroll up to the mapping section of the report and link to the map viewer through the mv link. Use the Display settings link to add the Contig and the GenBank map to the display. Use the zoom control graphic to zoom out until the entire contig is displayed. How large is this contig? How many GenBank records were used to construct it? How many of these are drafts and how many are finished records? What other genes are annotated on this contig? Examine the GenomeScan map in the region of BRCA1. GenomeScan has identified a gene in one of the introns of BRCA1. This may be a pseudogene for what human protein?
- One kind of hereditary hearing loss was recently mapped to a relatively small region on chromosome 6. The gene responsible appears to be between the markers D6S472 (also known as AFMa128yd9) and D6S1722 (also known as AFMa102ya5). Use the search option on the human map viewer to find both of these markers. Search with ‘D6S472 OR D6S1722’. Display your results by clicking the link under the chromosome graphic where there are hits. Remove all maps except the Genethon map and add a ruler using the display settings. Zoom in until you can approximate the distance between these on this Genetic map. How far apart are they? What are the units on this map? Replace the Genethon map with the STS map. This is the NCBI ePCR map. What are the physical positions of the two markers? Again find the distance between these markers. What are the units now? Adjust the region shown to display an interval just spanning these markers. (Enter the distances in thousand of bases, for example, 147,000K.) How many STS markers are in this region? Add the genes on the sequence map to the display. What identified genes are in this region? Link to LocusLink to see if this gene is now associated with autosomal deafness.
- There is set of sodium channel genes in the rat, that seem to have corresponding genes on chromosome 2 in human. A study by Escayg et al. used the rat cDNA (accession number M22253) for one of these rat sodium channels to find the corresponding gene in the human draft sequence with a BLAST search. They further showed that mutations in this gene were involved in an inherited neurological disease (generalized epilepsy with febrile seizures plus type 2 (GEFS+2)). Use the human genome BLAST page to search the draft human genome with the rat cDNA (M22253). How many contigs from chromosome 2 do you hit? Click on the link corresponding to the best BLAST hit and zoom out until you can see all of these contigs displayed. There are two sodium channel genes (SCN) annotated on the Gene_Sequence map; what are they? The gene identified by Escayg (SCN1A) and co-workers lies on the q terminal side of these. What contig contains it? Use the display settings to add the GenBank map. What accession number contains (SCN1A)? Is this draft (HTG) or a finished sequence? Note that there is also another as yet unannotated SCN gene between the annotated gene.
- The UniGene collection is a very useful resource for finding uncharacterized genes that are known only from ESTs. According to the release statistics, only a minority of the EST clusters contain a known gene. The majority then represent the transcripts of undiscovered, uncharacterized genes. We should be able to identify these unknown genes in the draft genome through a BLAST search. Starting from the UniGene page, retrieve the human UniGene Cluster Hs.333314. How many ESTs are in this cluster? Notice that the first EST (N93603) is a 3′ read. What does the “A” symbol next to this record read mean? There are some cases in this cluster where both the 3′ read and the 5′ read are from the same clone. Which ones are these? The non-EST sequences in this cluster are based on the National Cancer Institute’s Mammalian Gene Collection. This is a targeted resequencing of potentially full-length EST clones. Perform a human genome BLAST search with the MGC sequence from this cluster (BC006407). On what chromosome and in what region is the corresponding gene? On what contig is this gene? Look at the GenomeScan and EST maps for additional support for the presence of this gene. Find a nearby annotated gene.
- Glutathione-S-transferases (GSTs) are enzymes involved in a variety of detoxification processes including the metabolism of carcinogens. Polymorphisms in GST genes including the absence of certain genes have been associated with increased susceptibility to cancer. There is a cluster of Mu class GST genes on chromosome 1. Use OMIM in Entrez to find the entry for GSTM1. Use the link on the left sidebar of the OMIM entry to link to the OMIM gene map. What methods were used to place GSTM1 on the OMIM Gene Map? Follow the link from the OMIM Gene Map to the map viewer link to the map viewer. Use the display settings to remove the Morbid Map and the Gene_Cytogenetic map from the display and add the Variation (SNP) map. Make the SNP map the master map. How many polymorphisms are mapped for chromosome 1? Zoom in to an 8K region surrounding GSTM1. Do this by mousing over the Genes_sequences map and left-clicking on the graphic. A menu with various zoom levels will appear. You will need to zoom in multiple times. You can then adjust the range using the region shown boxes. There are a number of SNPs associated with the coding region of GSTM1. How many are there? (To see what the symbols mean on the SNP map click one of the RefSnp identifiers and link to the RefSNP Summary Info.) Two of these also occur in the coding regions of two other GSTM members on chromosome 1. Which ones are these?
- Use LocusLink to find the entry for the human glyceraldehyde 3 phosphate dehydrogenase gene. Click on the map viewer link ( mv) to find the map location and the contig containing the GAPD gene. Zoom in to see the exon-intron structure of the gene on the gene_seq map. How many exons are there? Now use human genome BLAST to verify the location and structure of this gene. Use the GAPD RefSeq (NM_002046) to perform this search. Set both the alignments and descriptions to 250. How many contigs do you hit in the human genome? Click on the Genome View button to see the distribution of these hits on the genome. Look at some of the high scoring single hits and to see what’s unusual about them. How can you account for these results?